Co-dependent Technologies (Test + Drug)
Presented at ARCS Conference 2024





Presented at ARCS Conference 2024
https://tacshealthcare.com.au/wp-content/uploads/2023/11/NSW-CC-Sep-2023-Poster-186.pdf
Objective
As one of the three pillars of Australia’s universal public health system, the Pharmaceutical Benefits Scheme (PBS) provides equitable access to medicines for all Australians. Cancer is a leading cause of death of Australians (18% in 2020).1 Hence, timely availability of new treatments on the PBS has become more critical than ever.
This retrospective review of the listing and use of cancer pharmacotherapies on the PBS considers whether the current scheme is meeting the needs of Australian medical oncologists, haematologists and their patients.
Methods
PBS and Repatriation PBS (RPBS) claims data for all funded programs, including chemotherapy, were sourced from the Services Australia 2 and PBS3 websites. Services (prescription dispensed for a PBS item) for and benefit statistics were graphed by Anatomical Therapeutic Chemical (ATC) first level code over time. Proportions of Government expenditure and average cost per service were calculated.
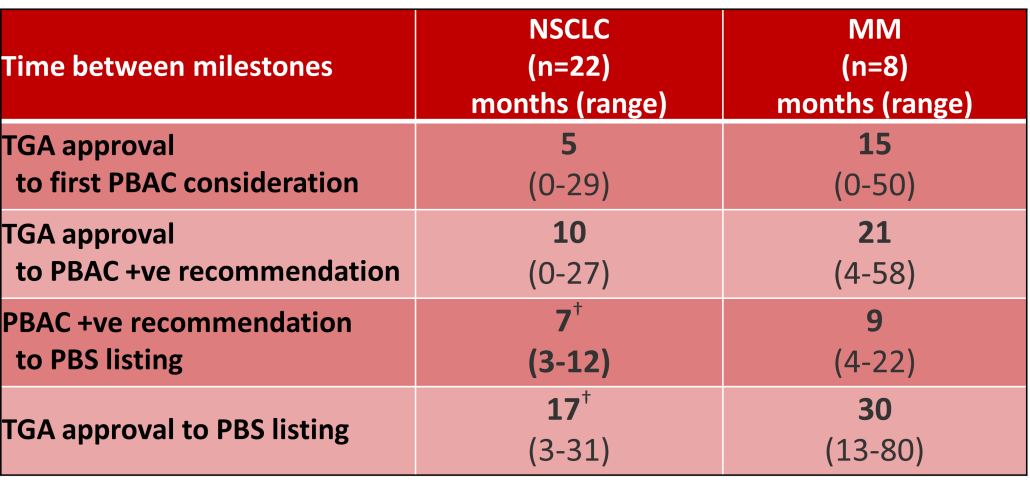
Outcomes of Pharmaceutical Benefits Advisory Committee (PBAC) Meetings4 were reviewed to identify considerations for the first indication of new products for non-small cell lung cancer (NSCLC) and Multiple Myeloma (MM), as representative of pharmacological treatments for solid tumour and haematology indications. The duration between marketing approval by the Therapeutic Goods Administration (TGA) and time points to PBS listing were determined.5
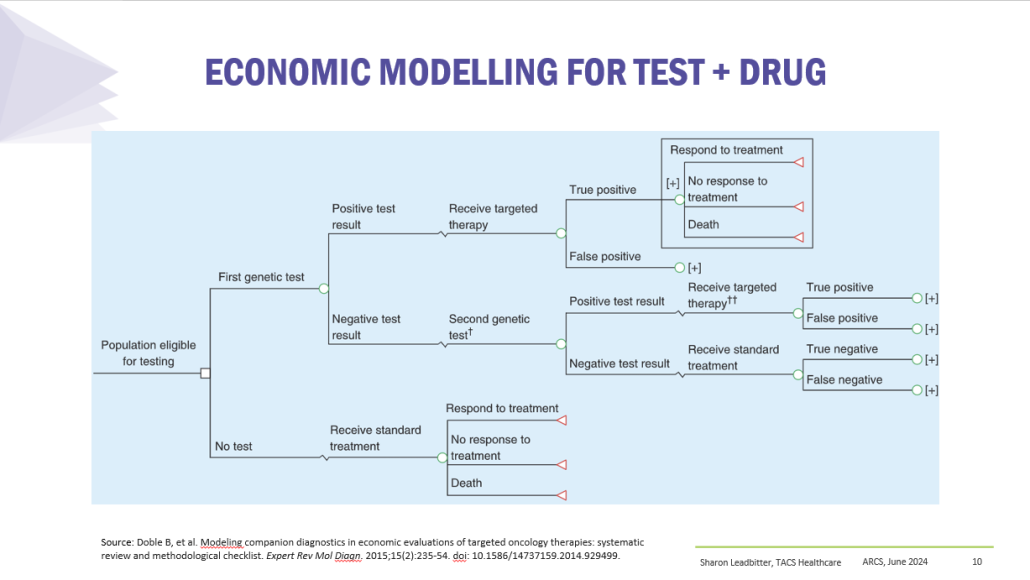
Results
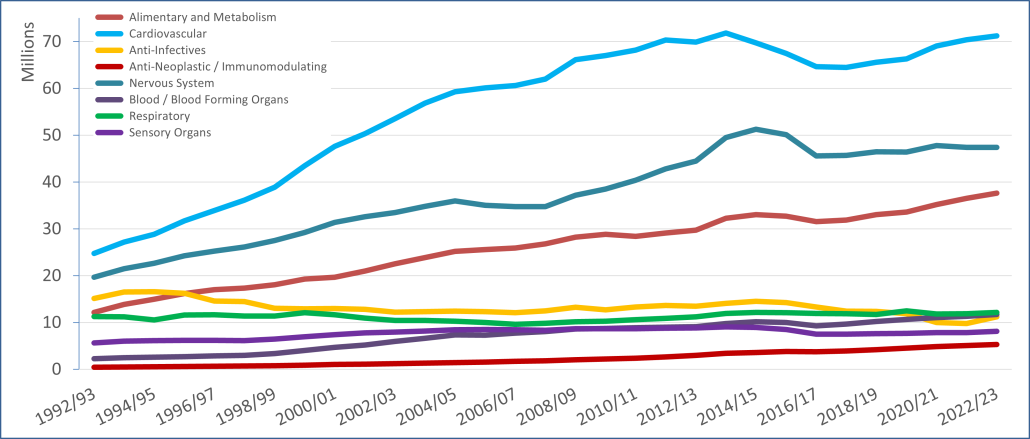
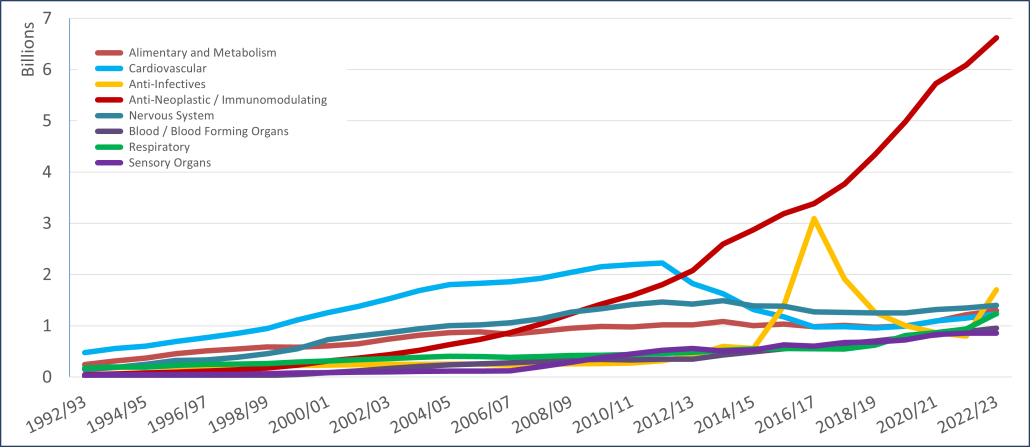
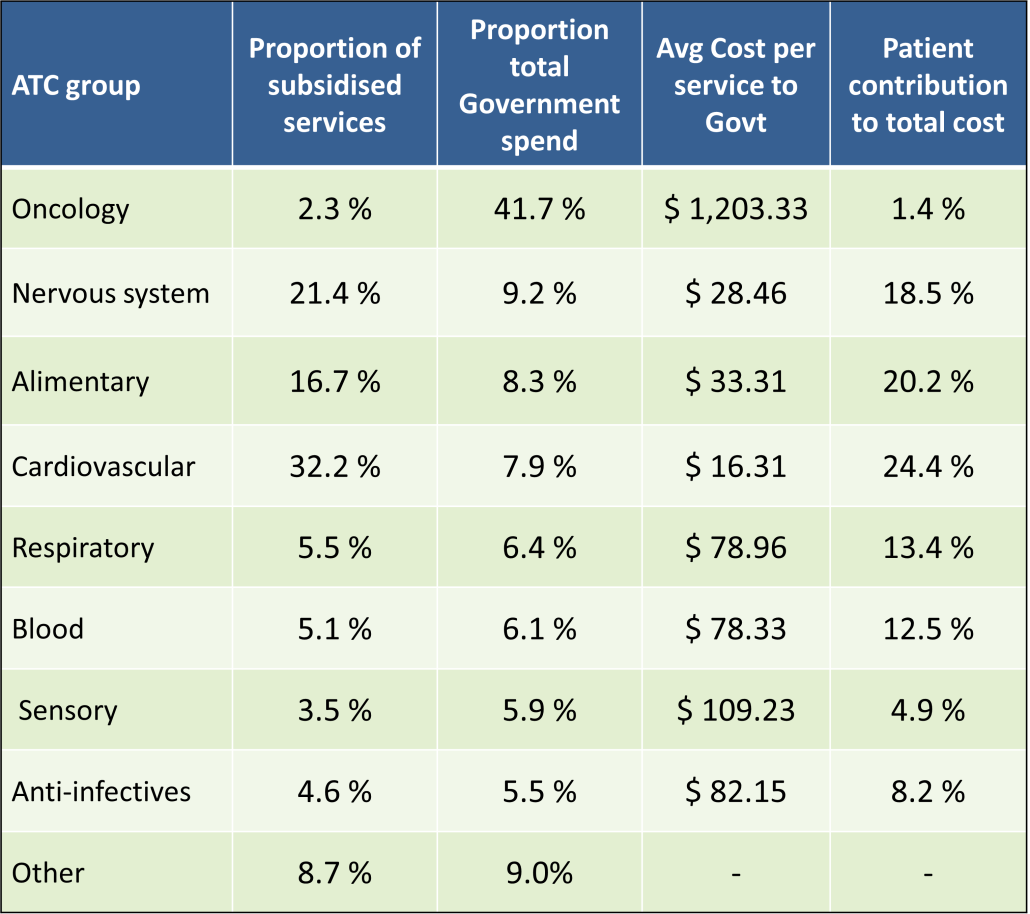
PBS/RPBS claims data was available by ATC for the 31-year period 1992/93 to 2022/23. Changes in the number of services provided and benefit paid by the Federal Government (excluding patient co-payments) for ATC groups contributing over 5% are shown in Figures 1 and 2. Table 1 provides cost breakdowns by ATC, including patient out of pocket contributions.
All ATC groups grew over time mirroring societal demographic changes. The higher growth of Cardiovascular, Nervous System and Alimentary groups reflect treatment innovations with statins, anti-depressants and proton-pump inhibitors, respectively.
The introduction of Section 100 High-Cost Drugs to reimburse States for in-hospital usage of certain therapies, and agreements between the Commonwealth and all States/Territories, except NSW/ACT, to permit PBS out-patient dispensing have also added service volume to the scheme. Increased costs have been offset by pricing policy changes, mostly directed at manufacturers, such as splitting into Formularies by molecule patent status, Price Disclosure and Efficient Funding of Chemotherapies (EFC).
While the proportion of total services for cancer treatments grew over time, the ATC group represented only 2.3% of PBS/RPBS activity in 2021/22. However, in the same year, the group represented 41.5% of total Government expenditure on the PBS/RPBS program and continuing to grow.
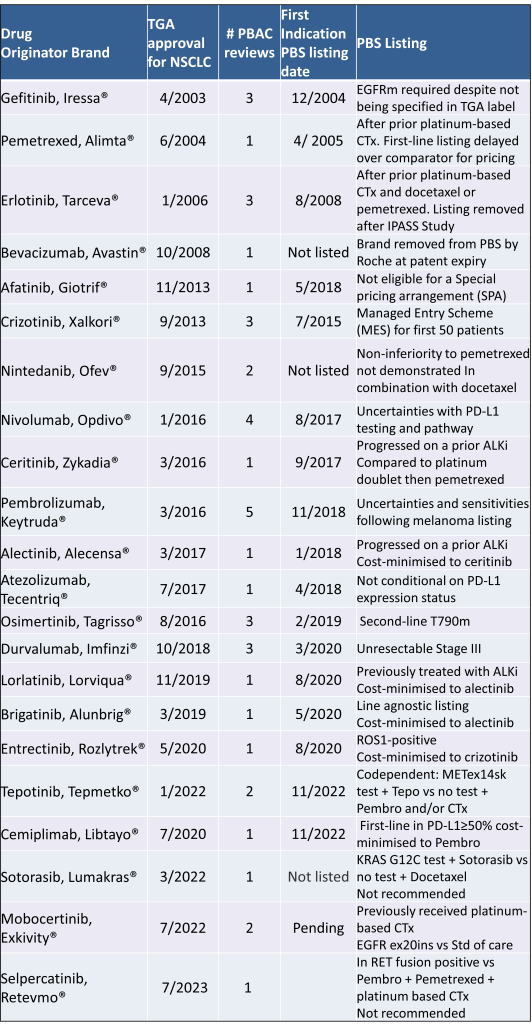
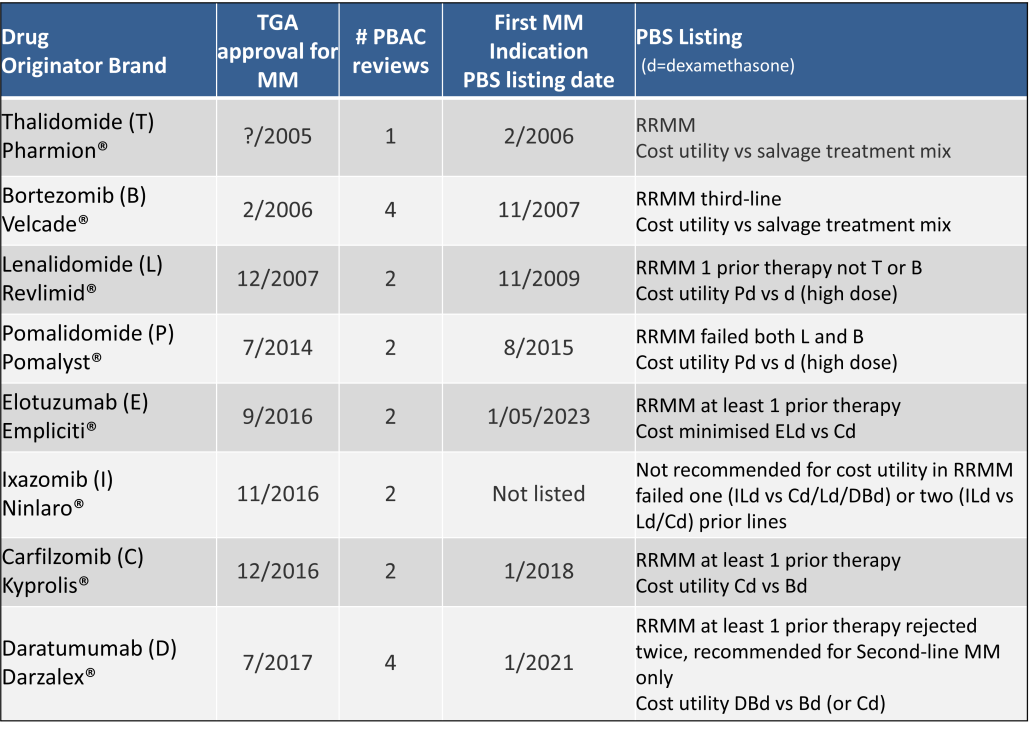
Table 2 lists new medicines requesting PBS listing for a NSCLC indication, and Table 3 for Multiple Myeloma from PBAC Meeting outcome documents available from December 1999. Public Summary Documents were introduced from July 2005. The number of PBAC considerations and date of TGA approval and PBS listing are noted.
Discussion
Innovative medicines are now considered by the PBAC at similar times to TGA approval due to policies such as Parallel Processing and TGA Provisional pathways. Streamlining of PBS processes in the past 5 years see clinically effective drugs navigating the process usually within two PBAC considerations. Beyond this, intractable issues can introduce a significant lag between local treatment practices and internationally accepted standards of care.
The finding that there was an average 13-month difference in time to access new treatments for MM and NSCLC on the PBS over the period supports ongoing policy reform to accelerate access.
Conclusions
•Although variable time to access, the PBS/RPBS is serving patients with cancer, and their clinicians by subsidising innovative treatments.
•Treatments for cancer account for an increasing proportion of total Government expenditure on the scheme. Currently, over 40% and growing.
•Although the proportion of patient out-of-pocket costs are comparatively low, the actual amount can be significant and inequitable relative to other therapeutic areas.
According to the Royal Pharmaceutical Society, medicines are responsible for ~25% of the UK NHS’s carbon emissions [1].
These emissions result from the manufacture, procurement, transportation, and use of medicines (20%), with the remaining 5% specifically from inhalers and anaesthetic gases [1].
On a global level, the health sector has been estimated to contribute approximately 4.4% of global net emissions by the Health Care Without Harm organization [2].
While simple changes such as opting to use propofol instead of desflurane as an anaesthetic reduces the total amount of greenhouse gases released over the drug life cycle [3], what consideration is given to the environmental impact of new medicines when being evaluated for pricing, reimbursement and access at launch?
With climate change happening at an ever-increasing rate, it’s no surprise that the topic of sustainability and environmental impact was raised in several forums at the recent HTAi Annual Meeting in Australia, as noted by one of our FingerPost consultants, Sharon Leadbitter, who was in attendance.
Is environmental impact a relevant dimension to include in HTA?
From our experience at FingerPost, the environmental impact is not a consideration for access, pricing and reimbursement decisions. Because of this, it is ignored in the value messaging, objection handling, economic modelling and HTA submissions for new medicines.
Payers do consider wastage and storage in relation to formulary listing decisions, but this is typically from a practicality and budget perspective, rather than an environmental one. But could this change in the future as sustainable prescribing policies are scaled up to regulators and/or payers?
Sharon conducted a targeted literature search via PubMed using key words ‘HTA’ and ‘environment’ to understand if environmental impact is currently being considered within HTA decision frameworks. The initial search produced almost 900 citations, of which the majority were irrelevant. The references from relevant articles published in 2023 were used to locate other publications and sources.
Here are her key take home points.
Environmental impact is not high on the agenda with current appraisal methods for new medicines…
Despite the increasing interest in sustainability of medicines, a 2023 review by Breslau [4] of 53 HTA guidelines for ten societal value elements† identified only one mention of ‘environment’. This was by the Second Panel on Cost-Effectiveness in Health and Medicine [5].
The Second‡ Panel recommended that the reference/base cases of all cost-effectiveness analyses should report from both a healthcare sector and a societal perspective [6].
Societal healthcare-related inclusions are costs of patient-time, unpaid caregiver-time and transportation to receive care.
Environment is included as a non-healthcare sector impact, along with productivity, consumption, social services, legal or criminal justice, education and housing [5] [7].
…despite European Regulation recognising the need for the sustainability of health systems
This aligns with Note 3 of the new definition of HTA developed though an international collaboration in 2020 [8], which says:
The dimensions of value for a health technology … include clinical effectiveness, safety, costs and economic implications, ethical, social, cultural and legal issues, organisational and environmental aspects, as well as wider implications for the patient, relatives, caregivers, and the population…
However, while the Regulation 2021/2282 of the European Parliament recognises the importance of the sustainability of health systems (EUR-Lex—32021R2282), it does not include the environmental dimension in the five non-clinical assessment domains of HTA [9].
Environmental impact could be a criteria within multiple technology appraisals, but it is not yet clear how to incorporate or assess
Guirado-Fuentes [9] conducted a scoping review of the literature considering the inclusion of the environmental dimension into HTA processes. They identified 22 articles published between 2010 and 2022 which could be grouped under four topics:
For further reading, see Toolan et al. (2023). Environmental impact assessment in health technology assessment: principles, approaches, and challenges [13].
Environmental Impact is not a key HTA/payer decision driver for now, but the growing pressure to reduce emissions is likely to move it up the health policy agenda over time
HTA and pricing frameworks are already complicated enough; adding yet another appraisal criteria is likely to slow decision making down and make it more resource-intensive for everyone.
Despite this, the impact of carbonization on health outcomes and increasing pressure from Governments to reduce emissions is likely to keep moving this up the healthcare policy agenda.
As this happens, environmental impact may become a key consideration in decision making alongside efficacy, safety and quality of life. There may be a need to justify why a less sustainable medicine has been listed on formulary, prescribed or administered where there is a like-for-like choice. It could, in turn, start to influence the standard of care or line of treatment. And this will then start to impact market access and commercialisation strategy, as well as acquisition and partnering deals.
For further insight, this topic could be expanded to a full literature review as there are a number of resources and thought leadership articles in the public domain on this topic. The Royal Pharmaceutical Society has published various Sustainability Policies in this area that provide further suggestions about how changes could be implemented by healthcare providers.1 McKinsey & Company also recently published an article identifying changes that MedTech and pharmaceutical companies could be making [14].
†defined as components beyond health impacts to the treated individual and costs beyond those incurred by the healthcare sector to deliver those interventions. The ten consumption, economic activity, education, environment, family spill over, healthcare system capacity, housing, legal, social services , transportation.
‡ The First Panel was the basis for the seminal book by Gold MR, Siegel JE, Russell LB, Weinstein MC. Cost-Effectiveness in Health and Medicine. New York, NY: Oxford University Press; 1996.
LinkedIn post, August 2023.
Many thanks to Sharon Leadbitter for researching and clarifying the current literature reporting on the Environmental Impact within HTA decision making.
Sharon is a Market Access specialist based in Sydney Australia with over 15 years biopharmaceutical company experience.
If you have any questions about this review or would like to understand how FingerPost can help you build and demonstrate product value ahead of exit or launch, please contact:
Catherine Bacon: cbacon@fingerpostconsulting.com
Nick Melling: nmelling@fingerpostconsulting.com
While the “Streamlining PBS Processes” work has completed, and the Strategic Agreement 2017-2022 of which it was a part, has passed into history, what is the ongoing impact?
The new classification of submission types became effective from the PBAC July 2021 Meeting (March 2021 cut-off). This meant the loss of the ‘major’ and ‘minor’ labels traditionally used to distinguish between those submissions requiring full evaluation and those managed at Committee Secretarial level. Definitions applying to both new submissions and re-submissions have been changed.
For new submissions, five categories apply based upon the complexity of the submission and evaluation required. The new processes also apply to vaccine listings on the National Immunisation Program (NIP).
Categories 1 and 2, formerly major submissions, require the PBAC to assess the magnitude of clinical improvement or toxicity reduction, the incremental cost and the comparative costs and outcomes where an economic evaluation is required to support a claim of cost-effectiveness, cost-utility or cost‑minimisation.
Categories 3, 4 and Committee Secretariat, formerly minor submissions do not require a full evaluation (clinical, economic and financial evaluation).
The chart shown includes submissions by category for formal and out-of-session PBAC meetings held in the 12 months from July 2021. This encompasses the three formal meetings of July 2021, November 2021 and March 2022; and three out-of-session, September 2021, December 2021 and May 2022. Unfortunately, despite 14 considerations at the May 2022 meeting, these were not classified during the new terms. For the purpose of analyses, these are assumed to be resubmissions.
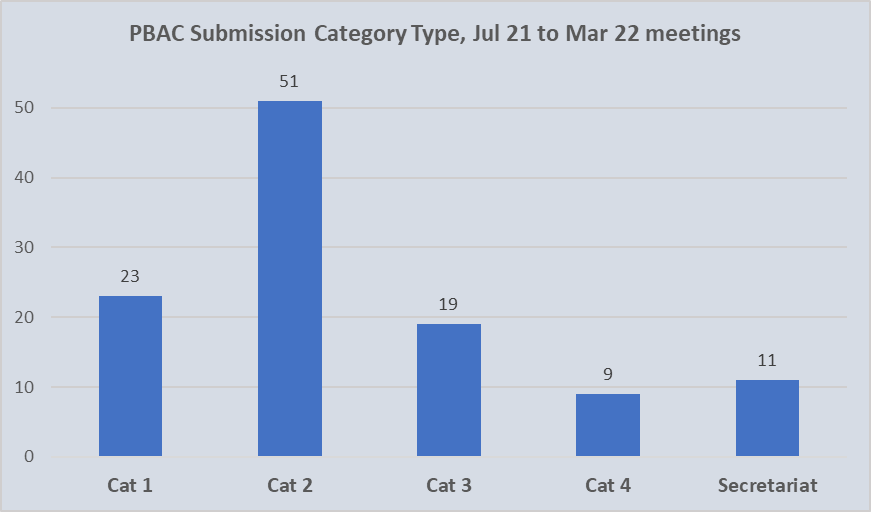
Of the 113 initial submissions categorised over the 12-month period, 74 (66%) were allocated as Category 1 or Category 2. Of these, approximately one third (23) qualified as Category 1 with an associated complexity or innovation. Given that overall, Category 1 submissions accounted for 21% of all those lodged, it does not reflect a decrease in novel pharmacological treatments being brought to Australia.
Interestingly, it is The National Health (Pharmaceuticals and Vaccines – Cost Recovery) Regulations 2022 that “prescribes the requirement for categorising submissions”. However, in relation to cost recovery matters, nothing takes precedence over the Cost Recovery Regulations.
Using the recently announced revised Cost Recovery Implementation Statement (CRIS), effective 1 August 2022, fees of approximately AU$ 15 million would have been received by the Commonwealth Department of Health and Aged Care over the 12 month period for new submissions. Fees for subsequent submissions and other services are not included in this total.
Category 2 is the default for submissions requesting a PBS listing that does not meet the criteria for Category 1.
Category 3 submissions relate to requests to change existing listings that do not change the population or cost-effectiveness of the medicine AND that do not meet the criteria for a Category 4 submission.
Although the PBAC will assess the clinical need for and clinical effectiveness of the requested listing, an economic evaluation is not necessary to support the claims made in the submission. Additionally, the financial estimates do not require the PBAC to assess any substantial financial implications for the supply of a listed medicine or designated vaccine.
They may also relate to a request for the PBAC to reconsider an existing recommendation where there is no change to the clinical, economic or financial information most recently relied on by the PBAC.
As PBAC advice is required on a case-by-case basis regarding the potential for schedule equivalence for biosimilar listings, Category 3 submissions are also appropriate for a new biosimilar brand of an existing pharmaceutical item with no indication changes.
Category 4 submissions involve a request for one or more of the following:
Committee secretariat submissions relate to applications where the requested listing changes do not require the PBAC to consider comparative effectiveness, cost-effectiveness or clinical need:
Note: Applications that do not require PBAC consideration for listing an additional brand (a generic medicine) or new oral form of an existing TGA-approved and PBS-listed pharmaceutical item should be lodged directly to the Department of Health &Aged Care.
Procedure guidance for listing medicines on the Pharmaceutical Benefits Scheme version 2.4
Since 1990, members of the ICH [1] have co-operated to develop guidelines and standards for demonstration of the efficacy, safety and quality of new products. As membership has grown, the guidance documents have been increasingly adopted as the international standard. Consistently satisfying the data needs of reimbursement agencies is yet to reach this level of uniformity. Will authorities agree on, and even mandate, inclusion of RWE into reimbursement submissions?

Reimbursement guidelines ensure that all relevant evidence published in the medical and grey literature is identified and presented systematically for evaluation. In the case of a new medicine, this usually is comprised of the clinical trial data generated for regulatory purposes. HTA landscape reviews across a number of therapeutic areas demonstrate that positive reimbursement decisions continue to rely almost exclusively on direct randomised controlled trials (RCTs) [2]. Exceptions are rare disorders and conditions of high clinical need when decisions are pragmatically made on the available data. Compromised reimbursement decisions may also occur when regulatory approval is given on the basis of single arm studies; and ethics approval for an RCT requires cross-over to be permitted after the primary endpoint.
The landscape reviews also show that the inclusion of analyses based on real-world data (RWD) definitely aid in reducing uncertainty around a decision. The price decrease required from what is “cost-effective” for listing being inversely proportional to the size of the arc of uncertainty. Uncertainty is significantly reduced when an included analysis demonstrates clinical effectiveness [3]; estimates local usage parameters and/or supports applicability of RCT results to the proposed patient population. However, this importance only applies only once a “yes” funding decision has been made.
This is why development of the reimbursement strategy for a new product must start early. In concert with regulatory plans, it is critical to identify the limitations/gaps in the clinical development program relative to the competitive landscape; to determine which type of data will most strengthen the evidence base; and to seek out alternate sources of data to generate this evidence in a timely manner.
The context for inclusion of real-world evidence (RWE) includes initial reimbursement discussions, pharmacoeconomic analyses, and conditional reimbursement schemes [4]. The right RWE can optimise restrictions or conditions associated with a listing, as well as the price achieved. This occurs at both a global and local level from data collection during early access programs, commissioning a clinical audit of hospital records, to requesting ad hoc analyses of a disease registry.

A snapshot of the status of RWE in reimbursement authorities:
Dr Hwee-Lin Wee from the National University of Singapore, presented a session on Real-World Data/Evidence use by Asia’s developing HTA systems during the ISPOR Asia Pacific 2020 Conference. The results of a 2019 survey confirmed that HTA agencies accept clinical effectiveness data from real-world sources as supplementary evidence to RCTs or where RCTs are lacking. Specifically, China, Indonesia, Japan, Malaysia, Singapore, South Korea and Thailand accept pragmatic clinical trials. China, Philippines, Singapore and South Korea publish specific guidance documents on use of RWD/RWE in HTA reviews [5].
This work contributed to the REAL World Data In ASia for HEalth Technology Assessment in Reimbursement (REALISE) working group. This is a collaboration between global experts and 11 Asian health systems. The group has developed non-binding guidance to provide a framework to generate and use real-world data (RWD) / real-world evidence (RWE) in a consistent and efficient manner for drug reimbursement decision-making in Asia [6].
During 2016, the Karolinska Institute, CHE York, University Departments from Argentina, Brazil, Columbia and Chile, ICON and Novartis collaborated to systematically evaluate the sources, characteristics and uses of RWE in South America. Following comprehensive literature and database searches and validation workshops during 2016, Justo and colleagues (2019) found that RWE was being utilised for pharmacovigilance and academic research purposes; little for HTA decision making and pricing negotiations and not at all to inform early access schemes [7].
The FDA has published guidance on the use of RWE to support regulatory decision-making for medicines and medical devices. These data may also support coverage decisions and to develop guidelines and decision support tools for use in clinical practice [8, 9, 10].
Lee et al. (2021) [11] conducted a retrospective analysis of the use of RWE in the cost-effectiveness analysis and budget impact analysis sections of final evidence reports published in the database of the Institute for Clinical and Economic Review (ICER). They identified 33 reports, all of which used RWE, most commonly for disease progression inputs (28.7%) and health care resource utilization and costs (21.1%). In 57% of cases, a retrospective cohort study design was used to collect the data, registry data was the most frequent (41%) data source, and 30% of RWE was industry-sponsored. RWE was rarely used for drug-specific clinical outcomes such as effectiveness (1.5%), adverse drug event rates (0.5%), and discontinuation rates (1.2%).
Malone et al. (2018) [12] interviewed 20 healthcare professionals from 18 different US health organisations about the use of RWE to inform Pharmacy and Therapeutic Committee decisions. Overall, RWE was considered useful, in particular to inform safety monitoring, utilization management, and cost analysis, but less so to guide coverage decisions. When it was used in decision making, pharmacy claims data was referred to by 100% of committees represented; medical claims (80%), Electronic Health Records (27%); Consumer surveys (20%) and Patient registries (13%). The usefulness of published RWE depended on the relevance and applicability, transparency of methods, study design and quality, and timing of any results. Perceived organizational barriers to the use of published RWE included lack of skill, training, and timely study results.
The FDA has also produced a number of guidance documents on communication with payors around RWE [13, 14] and between manufacturers and payors such as formulary committees [15].
In response to the need for consistency and integration of RWE efforts, a 4-year collaboration between researchers, payers, and patients known as the Canadian Real-world Evidence for Value of Cancer Drugs (CanREValue [16], https://cc-arcc.ca/canrevalue ) kicked off in 2017. Five working groups are developing and testing a framework for Canadian provinces to harmonise the generation and use of RWE for cancer drug funding. The aim is to build consistency in the use of RWE at a national level, which will lead to a robust pan-Canadian system supporting sustainability, value for money and improved patient care.
As part of this project Clausen and colleagues (2020) [17] conducted a qualitative descriptive study to understand current issues with the use of RWE in cancer drug funding decisions. The findings suggest that if RWE is to be used in drug funding decisions, there is a need for a cultural shift, improved data infrastructure, committed investment in capacity building and increased stakeholder collaboration.
The value of using RWE in regulatory decision making by the European Medicines Agency (EMA) is well established [18]. The inclusion of RWE in benefit-assessment/HTA evaluations continues to be the subject of numerous initiatives.
The LSE conducted a series of roundtable meetings in 2018 [19] to gain an understanding of the use of RWE across Europe and to assist the pharmaceutical industry to enhance their use of RWE. Three ways to advance were identified:
(a) prioritising the use of RWE by focusing on quality, including credibility of sources, design and methodologies,
(b) by identifying key areas or regions to pilot the use of RWE, and identifying supporting stakeholders, and
(c) by securing a consistent approach by developing an action bias, building best practice case studies, and demonstrating the value of RWE in contributing to decision making.
In a review of RWE policies of the HTA agencies of Sweden, UK, Germany, France, Italy and The Netherlands, Malady et al (2017) [4] found a lack of alignment in request for and acceptance of RWD in contexts of initial reimbursement discussions, pharmacoeconomic analysis and conditional listing schemes. Bullement et al. (2020) [20] found that the use of RWE in NICE submissions of cancer drugs was extensive, and appeared to have provided a valuable source of information to aid the decision making. NICE continues to proactively develop its use and acceptance of RWE. In 2019, a ‘Widening the evidence base’ statement set out NICE’s ambition to use broader sources of data and analytic methods to enhance existing methods and processes. Last year, NICE announced a collaboration with US based Flatiron [21] to explore how RWE can inform the clinical and cost effectiveness of health technologies.

The value of data collected outside of the clinical trial environment is clearly accepted by both payers and manufacturers with potential for use in evaluation and decision-making at all stages of the life cycle of a new medicine.
Representative bodies such as ISPOR and the International Society for Pharmacoepidemiology (ISPE) are collaborating to address the common issues noted as barriers to use of RWE. For example, recommending establishment of a register for RWE study protocols to improve transparency in research methods and analyses use. [22, 23] Numerous others continue to make valuable recommendations on how to move forward. [24, 25]
Policy consistency across organisations and jurisdictions is evitable. However, this will require moving beyond the established principles of evidence-based medicine into HTA 2.0, made possible by “big data” (see separate box). Additionally, new definitions of privacy and data security are required in an era where a search engine can know more about your health than your doctor.
Predictions are for a global market for sourcing, generating and providing RWD in a usable format of US$1.64 billion by 2024 [26]. As reimbursement authorities continue to receive more RWE of increasingly higher quality and value, it will become expected.
Image by Erika Varga from Pixabay
A recent study found no association between perfectionism and productivity at work. However, it did reveal that certain types of perfectionists are less popular with their colleagues. A “self-oriented perfectionist” sets very high standards only for him/herself; a “socially prescribed perfectionist” believes that the acceptance of others is dependent on his/her own perfection; while the “other-oriented perfectionist” expects flawlessness from those around him/her. No prizes for guessing which type people didn’t want to work with.
In a 2018 meta-analysis of perfectionism in the workplace, people were classified as either “excellence-seeking”, those fixated on achieving high standards, or “failure-avoiding”, those whose focus was on not making mistakes. This latter type of perfectionist were more likely not to be “agreeable”. That makes sense too.
According to table tennis champion Matthew Syed, in a podcast aired by the ABC, by avoiding mistakes we limit our opportunity to learn. Growth and new skills develop via the pathway of trial and error. Babies, infants and young children understand this as they learn the necessary skills to go to school. Unfortunately, despite all the advantages of an education, by the teenage years of peer pressure, failing becomes something to be avoided. As Syed points out, while the mindset to learn from mistakes is common in many sports, it is not prevalent in our social and political institutions.
Despite this intolerance for errors in institutions, Governments do make mistakes. By way of John Lord’s Great Australian Policy Stuff-ups series of articles published in 2019, I came across the Top-13 list. The list was compiled in 2006, so you will need to decide which more recent events surpass those included and summarised below. The full piece is an entertaining read.
13. Invention of Canberra (1908)
‘Canberra is a document of Australian immaturity‘, Sir William Keith Hancock (1898-1988), Australian historian, 1930.
12. Patrick White Wins the Nobel Prize (1972)
The Nobel Prize commendation said that White ‘for the first time, has given the continent of Australia an authentic voice that carries across the world’.
‘I am amazed at the way Australians have reacted, in a way they usually behave only for swimmers and athletes’, said Patrick White.
11. Federal money for science blocks at non-government schools (1963)
This was the beginning of Federal aid for private schools and the beginning of the end for the possibility of a student-centred system for funding schooling in Australia.
10. The Release of Cane Toads (1935)
In a sort of a reverse Stockholm Syndrome, cane toads have entered Australian popular folklore as ironic heroes. There was outrage when Federal MP Dave Tollner (from the Northern Territory) publicly embraced the killing of toads with a variety of blunt instruments, including golf clubs and cricket bats. A cane toad is notoriously difficult to eliminate by such methods, and not even backing a ute over one will guarantee its demise. Instead, the RSPCA recommends euthanasing it in the freezer.
9. Publication of John Stuart Mill’s On Liberty (1859)
Published in 1859, On Liberty is one of the foundation documents of modern liberalism. Its arguments for freedom of religion and freedom of speech were radical and enormously powerful. As the influence of Mill’s political ideas grew during the nineteenth century, more attention came to be given to his economic theories. The problem was that while in the political sphere Mill encouraged small government, when it came to economics, he embraced state intervention and industry protection.
8. The Labor Party Split (1955)
‘The Split’ kept the Liberals in power and kept Labor out of power during the 1950s and 1960s. Depending on one’s political sympathies, this was either a good thing or a bad thing. But the long-term effects of the Split were disastrous for both sides of politics, and for Australian politics generally. The root cause of the Split was communism.
7. Immigration Restriction Act (1901)
The very first Act passed in the new Parliament of Australia was to give effect to a White Australia Policy. The Immigration Restriction Act excluded non-white potential immigrants (and anyone else thought undesirable) primarily by introducing a dictation test whereby immigration officers could require potential immigrants to undergo dictation in any European language. Later, the dictation test was extended to any language. Racially based immigration was effectively abolished in 1966 when the dictation test was eliminated by the Liberal government of Harold Holt, and the Act was formally repealed by the Whitlam Labor Government in 1973.
6. WA Town Planning and Development Act (1928)
The Western Australian Town Planning and Development Act 1928 was the first such in Australia to give local government the power to control the use of private land. This had spread to all States by 1955 and increased in its regulatory intensity. The upshot has been a progressive and accelerating reduction in land available for housing. Most importantly, it has brought a vast increase in prices.
5. The Uniform Tax Cases (1942 and 1957)
There are two main villains in the story of the death of federalism in Australia. Commonwealth politicians should bear most of the blame—but their desire to centralise political power into their own hands is entirely to be predicted. The second villain, the High Court, is less easily excused. One of the greatest fallacies of Australian politics is the claim that the Constitution is difficult to change. What is usually forgotten is that a referendum is only one way of changing the Constitution. Another way the Constitution can be changed is by the decisions of the High Court, and successive Court judgements have completely subverted the balance between the Commonwealth and State governments.
4. The Montreal Olympics (1976)
Stephen Holland’s failure to win the Gold Medal in the 1500 metres freestyle at the 1976 Montreal Olympics was the key trigger for one of Australia’s greatest public policy disasters. From the initial setting up of the Australian Institute of Sport to fund some Olympic sports, Federal Government involvement has grown into massive public funding of elite sport across the board, with State governments joining in to set up their own facilities.
3. Wireless Telegraphy Act (1905)
The Wireless Telegraphy Act of 1905 inaugurated the century-long comedy of errors that is Australian media and telecommunications policy. Communications regulation also formed the sharp end of the wedge of big government in Australia. In 1901, the Postmaster-General’s Department was inaugurated with 16,000 public servants—90 per cent of the total Commonwealth administrative staff at that time.
2. The Harvester Judgement (1907)
The Harvester Judgement of 1907 effectively established the basic wage in Australia. The imposition of wage levels by judicial fiat, in defiance of prevailing conditions of supply and demand in the labour market, has been a disaster for Australia since that time.
1. The End of the Reid Government (1905)
The defeat of George Reid’s Government on 5 July, 1905 signalled the end of the last chance that Australia had to avoid the full imposition of the Australian Settlement. In the post-Federation parliaments, The Free Trade Party, lead by Reid, was the last to get a turn in government, following both the Protectionists, under Barton and Deakin, and Labor, under Watson. By putting the division on tariffs aside and conceding on other issues, Reid tried to get a joint Free Trade and Protection focus on ‘anti-socialism’. Some have argued that, if he had succeeded, free trade ideas would have remained a significant component of non-Labor politics.
Image: The Simpsons
True transparency requires more than making information available.
According to Rio Tinto, it was a ‘misunderstanding’ with the local Indigenous community that resulted in the destruction of 46,000 year-old sacred sites in May this year. The company had Government approval and stakeholders had been informed. All boxes checked. Obviously, that was not enough.
Since 2015, the Australian Government has been a member of the Open Government Partnership (OGP), committing to support the goals of increasing the transparency and accountability of government. The first OGP Global Report (May 2019, page 20), identifies these as generally lacking in health procurement decision-making in the 78 nations involved. In this article, I assume procurement includes the registration and reimbursement of therapeutic interventions by the Government on behalf of Australian taxpayers. My thesis being that openness may not necessarily be transparent, using two examples to illustrate.
Example 1. The presentation of options by the Therapeutic Goods Administration (TGA) for which type of medicines would have evaluation status made public.
Option 1: maintain TGA’s current publication arrangements [to not make public]
Option 2: list all applications accepted for evaluation
Option 3: list all applications at two different time points
Option 4: list applications of innovator medicines of highest public interest, but not generic or biosimilar medicines
The explanations given for providing Options 3 and 4 include: ‘Generally, there is less public interest in whether a generic or biosimilar medicine is under evaluation by TGA in Australia’, and ‘Earlier publication of generic or biosimilar approvals prior to ARTG entry allows more transparency of forthcoming competition to sponsors of originator medicines and potentially, purchasers of biosimilar and generic products.’
The TGA has been in the unenviable situation of neither being able to confirm nor deny whether a particular medicine is currently in the evaluation process. Other similar jurisdictions, such as Europe and Canada publish this information. It is a good initiative. The particular drivers of this 2019 consultation on transparency were: from the TGA side, inconsistencies with other agencies, especially evident during joint evaluations of new molecules; from the innovator sponsor side, more time to initiate legal proceedings against potential patent infringing generics/biosimilars and avoidance of liability for damages by the Commonwealth; and from generic/biosimilar applicants, maintain the status quo.
The inclusion of Options 3 and 4 show that there is an awareness of the potential negative commercial/financial implications to applicants and the Government of originator sponsors knowing the timing of registration of a generic or biosimilar. Is this made clear? Does ‘less public interest’ justify being selectively transparent?
Example 2. Measures associated with the ongoing Medicines Australia-Commonwealth Strategic Agreement PBS process improvements include the PBAC preference for greater transparency to be introduced in Public Summary Documents (PSDs) through a standardised approach to redactions.
Objectives of proposed PSD Changes – PBAC (Medicines Australia, Feb 2020)
•Increase transparency of PBAC’s decision-making processes.
•Publish PSDs in an efficient and consistent manner through establishment of a standardised approach to redacting information.
•Provide consumers with access to information to assist with making decisions about their individual health needs.
•Increase the public’s understanding about PBAC decisions.
•Align Australia’s practices with leading international jurisdiction approaches*.
*This refers specifically to the additional, and unexpected, push for PSD inclusion of all clinical data provided in PBAC submissions, irrespective of the publication status nor commercial impact. While no other jurisdiction currently requires it, other countries are considering in response to calls for increased pricing transparency (WHO, WHA, 2019).
While appropriate that the PBAC want to clearly indicate how each recommendation was reached, what is the point of providing even more information in PSDs when the existing format is, realistically, inaccessible to anyone without disease and technical knowledge and an appreciation of the meaning of uncertainty in this context? Inclusion of a summary in lay-man’s language in PSDs would go much further towards openness. It is like providing a patient with a copy of his/her CT scan and the report without an explanation as to what it means.
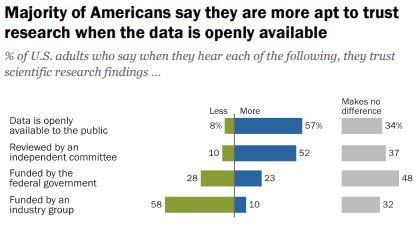
Do we want people to be able to understand the information OR is the fact that it is openly available more important?
According to a 2019 Pew Research survey, it is the later. Americans confidence in scientific findings are most influenced by open public access to data and independent committee reviews. There appears to be no similar survey providing Australian opinions.
Scientific journals are strongly encouraging authors to make datasets open source at the time of study publication with the intent of it becoming a mandatory requirement. Should sponsors of reimbursement submissions expect anything different? It is notable that journals target technical audiences, while PSDs are ostensibly for consumers (patients, healthcare professionals, competitors).
Before public release of information, consideration should be given as to who will use it, for what purpose(s), and the most appropriate release format, which is what the TGA and PBAC have undertaken via consultation in the examples provided. Is it enough?
Note: There is an opportunity to voice your opinion on ‘What does open government mean to you’ as input to the Third National OGP Australian Action Plan 2020-2022.
Image source: Jim Pavlidis
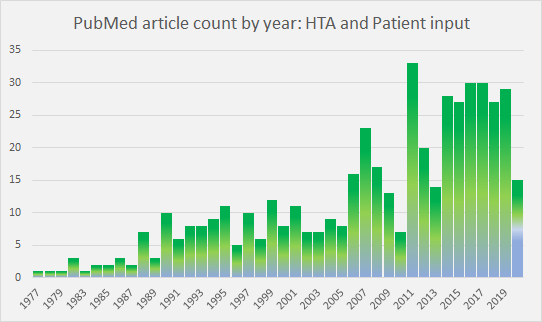
Patient input to HTA decision-making as a subject in the literature has peaked and plateaued in recent years (Figure 1). As a consequence of research and initiatives, such as those noted at the end of this article, the rationale for patient involvement in HTA is now well understood.
However, there is still a way to go in implementation.
Wale and Sullivan (2020) explore how patient input was reflected in the final documentation of HTA decisions by NICE, SMC and CADTH for a chronic disease and a cancer treatment considered during 2016-17. They discuss the influence of where in the process input is sought; how it is communicated (directly or written); by whom (individual patients or patient advocacy group); as well as the content of (clinical or realities of the disease beyond data); on the level of impact achieved. These and other opportunities to better involve patients in the HTA process were identified by Biointelect during research and interviews with NICE, SMC and CADTH, as well as PBAC stakeholders, for the BMS commissioned ‘Broadening the Evidence Report‘ (June 2019) (Figure 2).
In terms of the PBAC, Public Summary Documents (PSDs) currently include ‘Consumer comments’ in the Section titled ‘Consideration of the evidence’. The number and source of submissions received are noted, usually along with a summary of key points and a comment on usefulness to the Committee. No direct feedback is given to those who provide input. This may change with the most recent PBS Listing Procedure Guidance (V 1.7, April 2020) noting that the Consumer Input process is ‘under review’. The 2019 establishment of a designated ‘Consumer Evidence and Engagement Unit‘ in the Department of Health to ‘focus on expanding opportunities for consumers and patients to be central to ensuring that robust decision making can also support better transparency and understanding of HTA decision making processes‘, is likely involved in such changes. The recently launched Medicine Status Website was the product of considerable collaboration within and outside the Department.
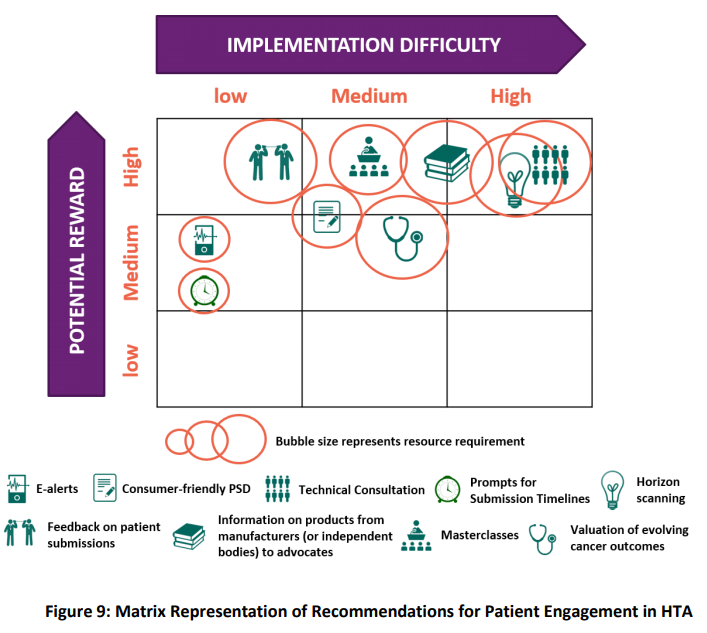
Street and colleagues (2020) say the terminology used to describe community participation in HTA, ‘is contested and frequently confusing‘. Depending on the jurisdiction, the terms: patients and consumers (with and without the advocate suffix), public, lay members, customers, clients, and citizens are used, often interchangeably. The article identifies the lack of a specific definition of the ‘public’ in the context of HTA, and propose the following:
‘A community member who holds the public interest and has no commercial, personal, or professional interest in the HTA process.’
This leads to the question, are ‘public’ and ‘patient’ engagement currently being viewed as the same thing during HTA processes? The patient perspective is lauded as it provides insights that clinical data often does not. That is invaluable to decision makers. However, where is the public voice, or more importantly, public values in the decision? Street (2020) notes that there is little empirical evidence on which to determine how different patient and public values are in this context. Patients may have a higher tolerance of risk, and, understandably an interest limited to a specific population and treatment. The public preference is towards equitable distribution amongst all patient groups, and the utilitarianism aim to maximise the well being of all.
In acknowledging this potential difference, UK’s NICE has established a Citizens Council. The Council is made up of 30 members of the public representative of UK demographics. It meets once a year and provides a perspective on relevant moral and ethical issues. The Council’s recommendations are incorporated into NICE principles and, where appropriate, into NICE’s methodology.
In the absence of the public perspective, do we assume that consideration of public values in HTA-based decision-making is the role of elected officials, in this case, the Minister of Health and Cabinet, who are the ultimate arbitrators of how to spend taxpayers funds? This will become increasingly important to understand as the pressure of unsustainable demands, driven by an ageing population and new technologies, require further rationing of healthcare costs.
The current review of NICE is a useful foil upon which to consider the announced refresh of the Australian National Medicines Policy. Both are being driven by concern that access processes are not keeping pace with biomedical innovation.
The table below provides a side by side comparison of the two appraisal systems and HTA reimbursement environments. Of interest is the relatively recent increase in NHS responsibility and focus on overall budget impact of reimbursing a new technology.
The need to effectively manage uncertainty whilst still making decisions is a key challenge of providing timely access, especially as patient populations are becoming smaller and more targeted.
 | ||
| Established | NICE became a legal entity in April 1999. First guidance published was an assessment of zanamivir for flu. | The PBAC evolved from the Formulary Committee and became an independent statutory body under s 101 of the National Health Act (NHA) in 1953. Cost-effectiveness was introduced into the NHA in 1998, with the first PBAC Guidelines published in 1992. |
| Responsible for | Technology appraisals (TAs) that assess the clinical and cost effectiveness of health technologies (pharmaceuticals, biopharmaceuticals, procedures, devices, diagnostic agents) and highly specialised technologies (HST). | The primary role of the PBAC is to recommend new medicines for listing on the Pharmaceutical Benefits Scheme (PBS), taking into account the medicine registration, its clinical effectiveness, safety and cost-effectiveness compared with other treatments. |
| Cost Recovery Fees | New regulations came into effect 1 April 2019 allowing NICE to charge for appraisals. A single TA costs approximately US$ 156 K for large, and US $40 K for small companies. | Significant increases from 1 July 2019, with the full process to listing, including one major submission, costing approximately US$ 300 K. A second stage is to be implemented from July 2020 but may be delayed. |
| Current review | Review of Methods and Processes Announced July 2019 | Federal Government review of the National Medicines Policy. Formerly announced October 2019 |
| Review Scope | Evaluation methods of technology appraisals and highly specialised technologies (HST). The QALY as a decision tool is not included with NICE indicating that changes to methods will only occur if there is a compelling case. | Consider whether the policy in its current form continues to meet the needs of Australians. Task force established, Terms of Reference pending. On-hold as resources directed to COVID-19. |
| Stakeholders | Substantial interest from external stakeholders and interest groups. | Wide-spread interest and involvement of health stakeholders, patient and consumer groups. |
| Review Concerns | – access and impact on patients -affordability in the NHS -clearer criteria for HST -improved representation of the patient view -inclusion of non-health benefits -success of the life sciences sector | -timely and equitable access to new, innovative therapies -affordability (Govt, community, patient) -supply chain (shortages, rebates) -integration of patient voice -viable medicines sector |
| Average evaluation | 16.0 months (2009 – 2016) | 4.5-month cycle from submission to PBAC consideration |
| Average time registration to reimburse (2012-2017)* | 4.2 months | 13.8 months |
| % NCEs registered, subsequently reimbursed (2012-2017)* | 84.3% (29% not funded for full licence, and 5% only recommended for the Cancer Drugs Fund). | 46% |
| Pricing | Voluntary Pricing and Access Scheme (VPAS) came into effect in January 2019, replacing the Pharmaceutical Price Regulation Scheme (PPRS). VPAS promises more and faster NICE appraisals for NCEs and speed up of appraisals for non-cancer medicines to be in line with cancer medicine timelines. | Following loss of the Pharmaceutical Benefits Pricing Authority (PBPA) in 2014, the Department of Health has become the sole arbitrator of pricing. Reforms in 2015 introduced statutory price cuts at 5, 10 and 15-year anniversaries from PBS listing targeting patented medicines. |
| Last major review | Introduction of end-of-life criteria and formation of the Cancer Drugs Fund (CDF) that relaunched under NICE in 2016. | Strategic Agreement 2017-2022 – Streamlining PBS Processes in progress. PBAC Guidelines Version 5.0 published September 2016. |
| Parliamentary level reviews | An inquiry by the All-Party Parliamentary Group (APPG) on Access to Medicines highlighted demand for wider value thresholds and modifiers, better management of uncertainty and improvements in the use of data and real-world evidence be brought into the methods. | Senate Inquiries into the Supply of Chemotherapy Drugs (2013); Availability of New, Innovative and Specialist Cancer Drugs in Australia (2015) and Funding for Research into Cancers with Low Survival Rates (2017). Recommendations accepted and implemented by Goverment to varying degrees. |
| US trade deals | UK-US post-Brexit trade agreement under negotiation. While US propose to redesign the NICE process, NHS England has been taking steps to increase its role in affordability and to effectively bypass NICE when appraising innovative medicines. NHS will play a key role in applying pressure on drug prices irrespective of any trade deal. | 2005 AUSFTA was instrumental in introducing major reforms to PBS listing process. Many, such as Independent Reviews and Public Summary Documents have been diluted, or configured to suit DoH purposes. For example, recent call for 100% transparency of clinical data. Others, such as PBAC hearings, continue to be invaluable. |
| Budget impact (Ghabri and Mauskopf 2018). | NHS Commercial Medicines Unit responsible for Patient Access Schemes and negotiation of outcomes-based pricing agreements. Introduction of the budget impact test makes a high budgetary impact the reason for manufacturers to reduce their price, either directly or indirectly, by lowering the cost-effectiveness threshold. | Since 2010–2011, any recommendation by PBAC that has a financial impact for the Federal Government is considered by the cabinet. The estimated financial impact of a drug on the Australian drug budget is a significant predictor of the PBAC recommendation for reimbursement |
| Managed Entry | Review will consider how to better managing uncertainty around early data and surrogate endpoints, such as collecting RWE while trials pending. | Process available since February 2011. Infrequently utilised following outcomes of initial agreements, . |
| Political Interference | Health Secretary Matt Hancock’s recent intervention in funding decisions around cystic fibrosis drugs, Orkambi and Trikafta, shows that NICE’s independence can be compromised. | Parliamentarians exert pressure on the Minister of Health (MoH) to enact listings once a positive PBAC recommendation has been made. However, MoH can deflect back onto Sponsor for not meeting the advice (conditions) of the PBAC recommendation. Strong commitment to this ‘independence’ of the PBAC. |
| Role of consumer and patient advocacy groups | The Review proposes new responsibilities for external stakeholders, such as delivering quantitative evidence of disease modifiers to be used in assessments and decision making. Disparity of resource and HTA skills within the patient group community to address these issues could risk furthering the inequality and lead to a misrepresentation of some therapy areas. | Increasing call, and opportunities for patient voice to be heard during evaluation of new medicines. Pilot initiative whereby patient groups meet with PBAC members prior to consideration of a relevant submission, is to be formalised. Industry increasingly engage with patient groups, especially around relevance of clinical endpoints and capturing real impact of disease. Government focus on transparency has lead to development and launch of a publicly accessible Medicines Status Website. |
References and Notes:
Lanning R. Does NICE hold the cards to drug pricing and reimbursement? PharmaField 30 March 2020.
Boyd N. A NICE Transformation? PharmaTimes Magazine, April 2020, p. 25-26.
*Medicines Australia 2018 COMPARE 4 Report
Add periodic pandemics to the ageing populations, accelerating rates of chronic disease, innovative technologies and price increases that are already challenging the durability of healthcare systems and the phrase, ‘a perfect storm’ comes to mind.
Prior to this recent challenge*, many countries have managed their health care system budgets by using a variety of prioritisation methods:
The pressure on budgets will escalate prioritisation decisions and as the impact is felt by more people, there will be public concern. Governments must proactively engage patients and the general public in the process. To date, those directly affected by access decisions become involved. This base of public participation needs to broaden to ensure continuing societal agreement with how decisions on spending finite resources are made. Understanding this, Littlejohn and colleagues (2019) have developed an online decision-making audit tool as a way to interact with stakeholders and help generate acceptance for the need for health prioritisation and the resulting decisions.

As parodied in the episode of ‘Yes Minister’, The empty hospital (BBC 2007), a hospital with no patients is extremely efficient to run. However, placing a greater focus on preventative health is a better way to reduce demand on health care systems. This could include initiatives to provide education to improve population health literacy combined with Government services focused on the social and environmental determinants of health and incentives for individuals to make responsible health choices .
Health care is big business. There are many vested interests who will lobby hard to maintain or improve their position. Incentives will also be needed to encourage identifying opportunities to improve efficiency and eliminate waste, a priority for those currently in the system. The poor take-up of Health Care Homes is an example of how a good idea can be derailed.
At that same breakfast, Professor Andrew Wilson ‘said the crunch point that would finally prompt action on health care reform would come when it was recognised that Australia’s states, which have very limited revenue raising capacity, could no longer afford to run hospitals. He was also concerned he said, about the affordability of healthcare for consumers, with rising out-of-pocket expenses, and a two-tiered health system that saw insured consumers able to access more extensive health care through the private system than those who used the public health system, even though 60+% of private health care costs were funded directly or indirectly by the public purse.’
*A prophetic report published last January by the World Economic Forum and Harvard Global Health Institute calculated that the annualised costs of flu pandemics alone are similar to those predicted to be caused by climate change. It recommends businesses (and Governments) to prepare to mitigate the impact of pandemics with the same urgency as they are for climate change.

{kind=link}